Our research in Computational Systems Epigenomics is primarily aimed at trying to understand how aging and exposure to cancer risk factors predisposes normal cells to carcinogenic transformation. In particular, we hypothesize that DNA methylation changes may play a key role in the earliest stages of cancer development, in driving epigenetic reprogramming and cellular plasticity, two key precancer hallmarks. Thus, a key aim of our research is to elucidate the role of DNA methylation changes in aging and cancer.
Our approach is both theoretical, recognizing that biology is not an isolated system but a complex system, drawing on advanced concepts and methods from network physics, signal processing and increasingly also artificial intelligence, as well as pragmatic, in order to meet urgent clinical needs in the cancer risk prediction and prevention field.
Broadly speaking our current research falls within the following research areas:
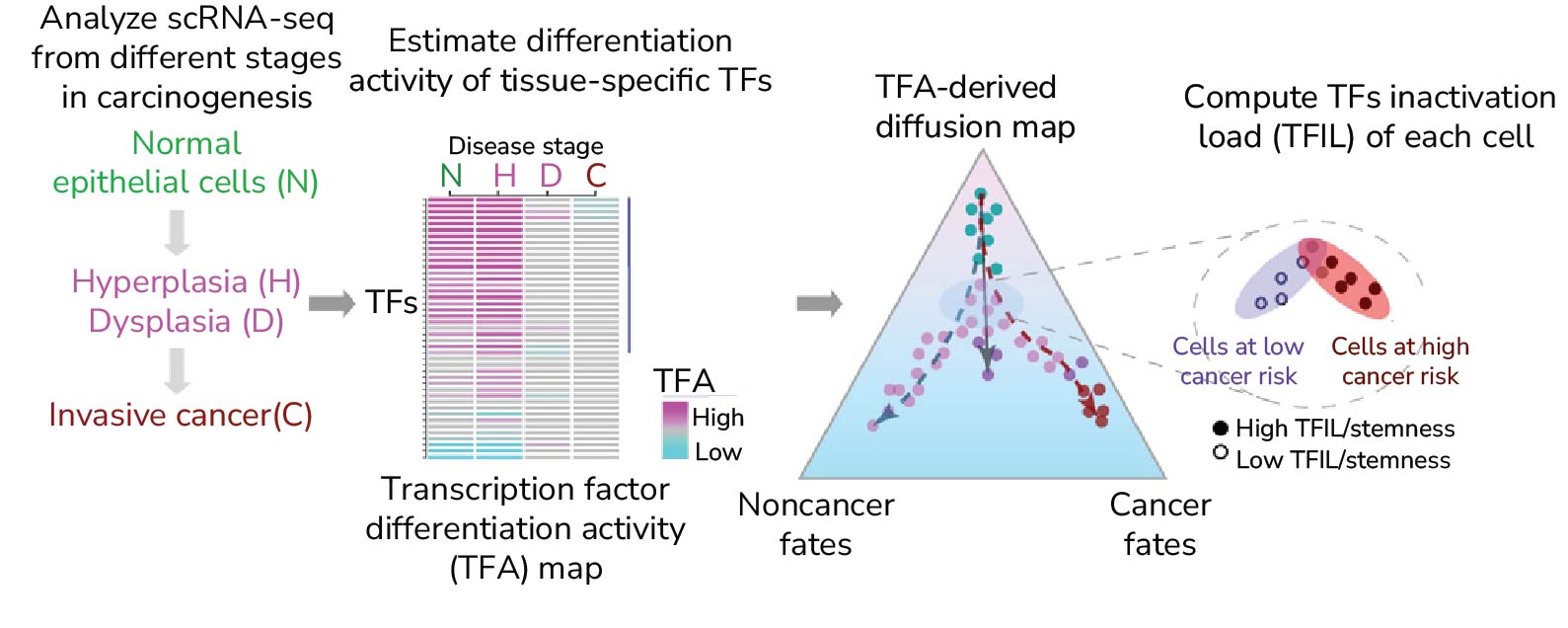
We are interested in using single-cell data (scRNA-Seq/snRNA-Seq) to elucidate systems biological principles of oncogenesis and to develop cancer risk prediction strategies at single-cell resolution. We have made substantial progress in coming up with the CancerStemID algorithm that allows identification of those cells in preneoplastic cell populations that are at higher cancer risk. Our collaborative work supports the view that DNA methylation changes are associated with epigenetic reprogramming and increased cellular plasticity in preneoplastic cell populations. From a more theoretical perspective we are exploring statistical mechanical approaches to building Waddington-like landscapes of the preneoplastic state, which could inform us about which cell-types are more likely to turn cancerous
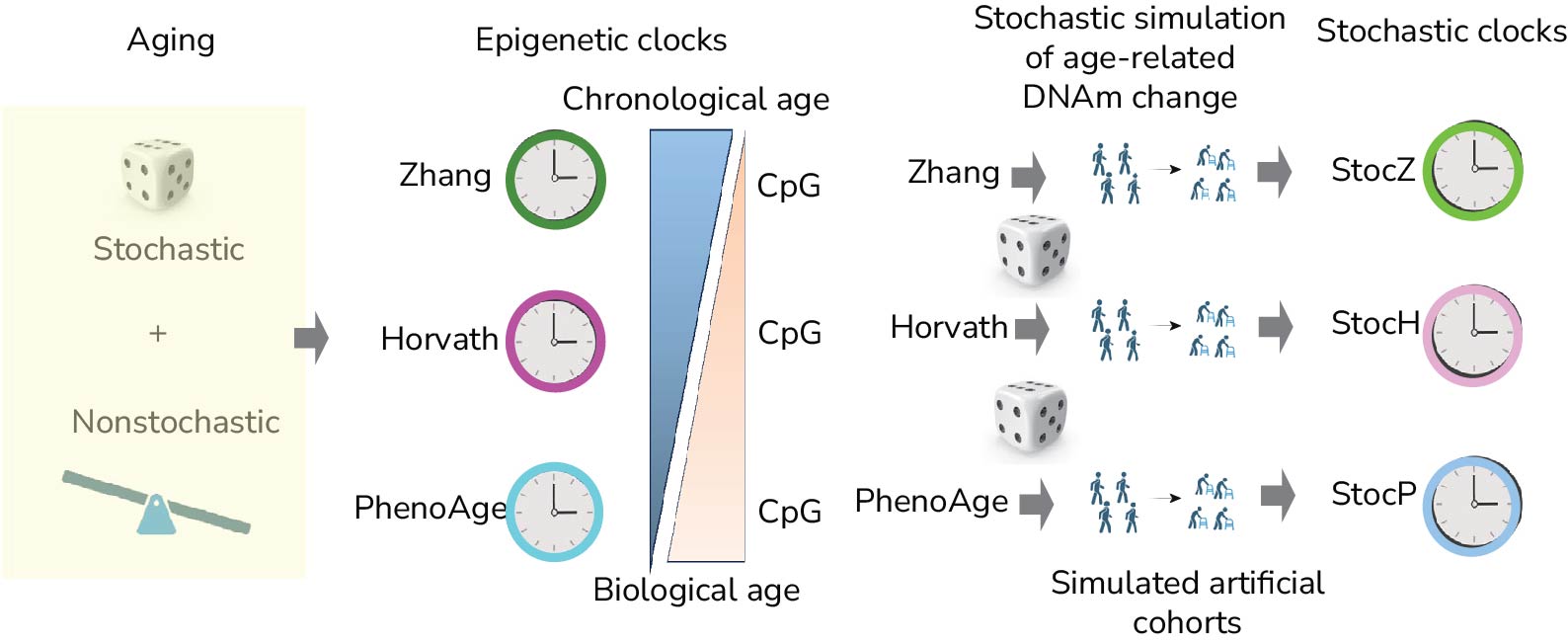
Epigenetic clocks have emerged as promising aging biomarkers. These and other tools may allow identification of individuals who are aging faster than normal and who are hence at higher risk of developing disease. We are particularly interested in understanding and decoding the DNA methylation landscape in aging, specially in relation to the molecular mechanisms that make these epigenetic clocks tick. In recent work, we demonstrated how stochasticity at the single-cell level could underpin the accuracy of an epigenetic clock to predict chronological age, with non-stochastic process driving biological aging. Another main focus has been on cell-type heterogeneity, which presents statistical challenges to interpretation, and which we are addressing by building cell-type specific clocks. We are currently exploring new improved strategies for building such cell-type specific epigenetic clocks. Another line of investigation is based on using concepts and ideas from theoretical physics to study aging and to develop novel aging biomarkers
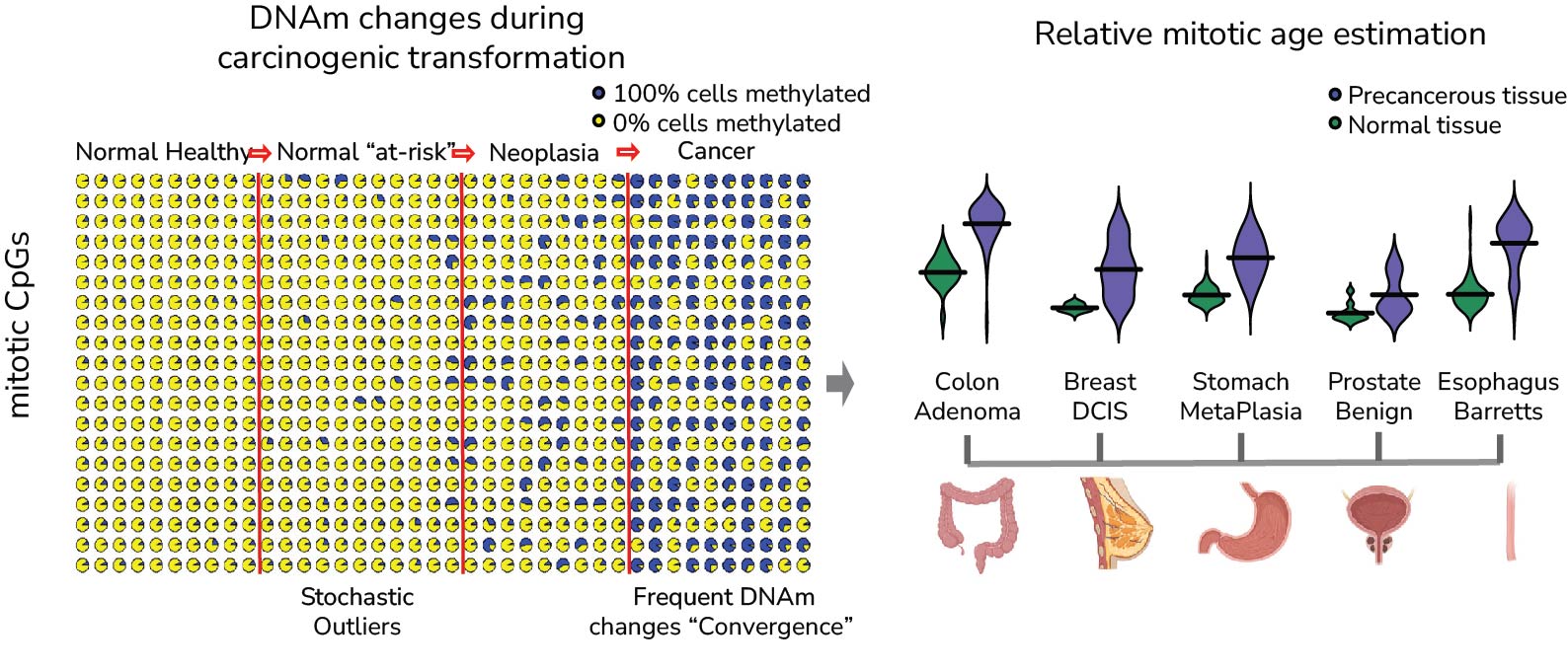
DNA methylation changes accrue in normal cells as a function of cell-division and have been shown by us and others to be reliable trackers of mitotic age (the epiTOC, epiTOC2 and stemTOC clocks). Mitotic age can be broadly defined as the cumulative number of stem-cell divisions in a tissue. Interest in quantifying mitotic age derives from the observation that it constitutes a major cancer risk factor. Hence, in principle, mitotic clocks could be used for cancer risk prediction. However, here too, one main challenge is cell-type heterogeneity since single-cell DNAm data is still too sparse and costly, with hence most DNAm data having been generated in bulk-tissue which is a complex mixture of many cell-types. Thus, we are developing statistical methods to build cell-type specific mitotic clocks in the hope that these can deliver more accurate predictors of cancer-risk.
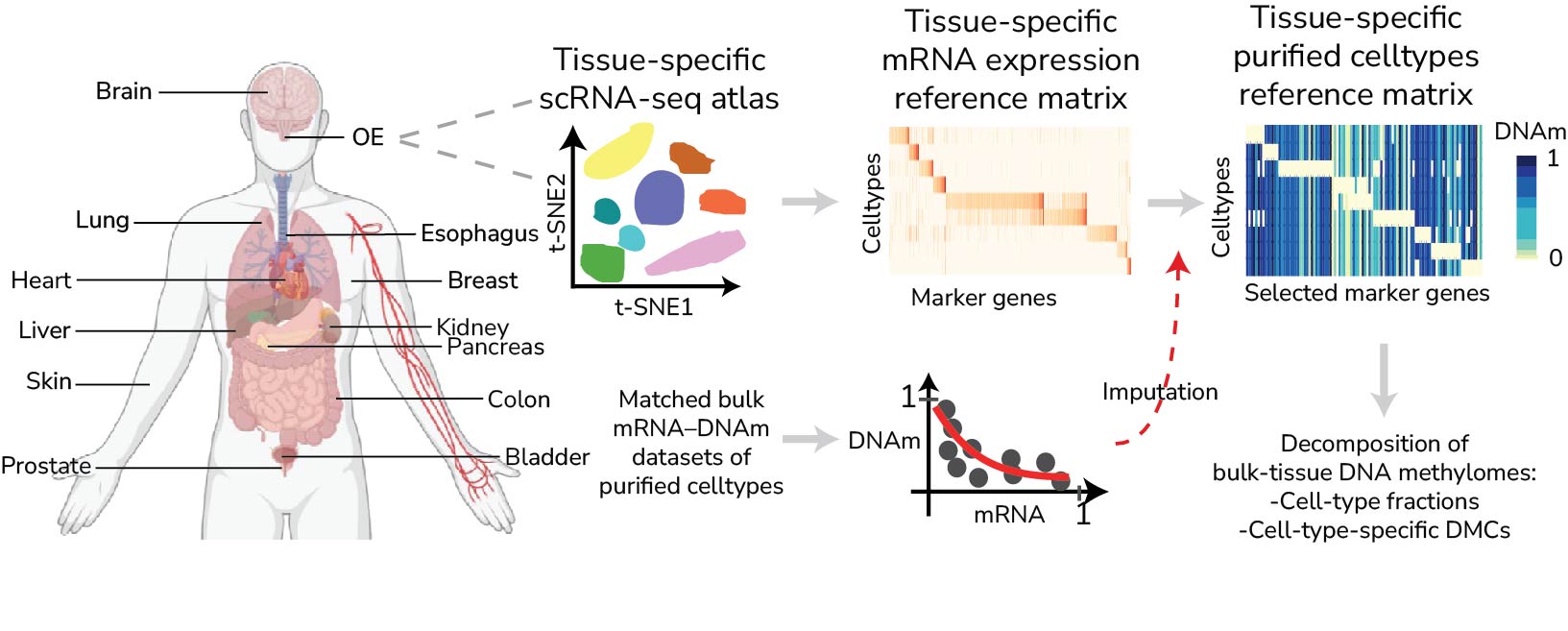
Given the significant challenge cell-type heterogeneity presents to the analysis and interpretation of DNAm data, over the years we have spent substantial effort in building cell-type deconvolution algorithms, not only for blood, but also for arbitrary solid tissue-types. In particular, we built the EpiSCORE algorithm and associated database of DNAm reference panels for 13 tissue-types. We are currently working on developing an improved version of EpiSCORE, as well as generalizing this database to additional human tissue-types, as well as to mouse, to allow cell-type deconvolution in solid tissues from mice
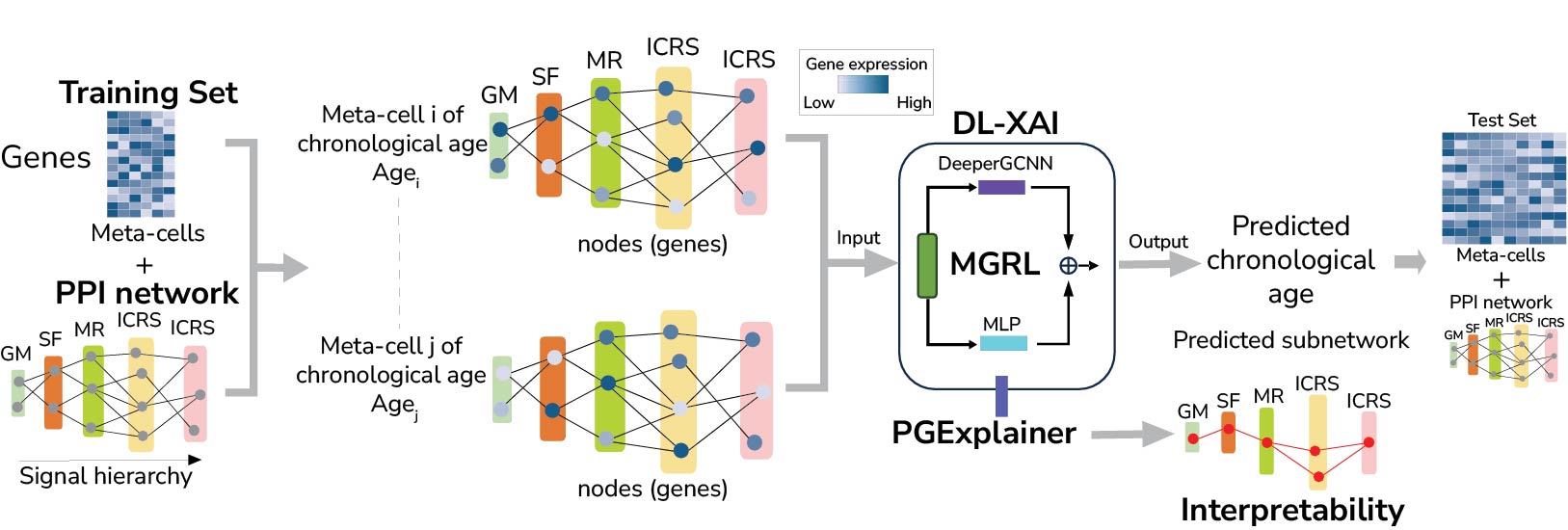
In order to better understand how aging alters normal cellular function, we have developed machine learning methods to help elucidate systems biological aspect of the aging process. For instance, we used a transcription factor (TF) regulon based approach to generate the first map of age-associated TF activity changes at cell-type resolution in mouse, using scRNA-Seq data from the Tabula Muris Senis. We have also explored using graph convolutional neural networks to obtain novel insights into aging at both the mRNA and DNAm levels. We are continuing these lines of investigation
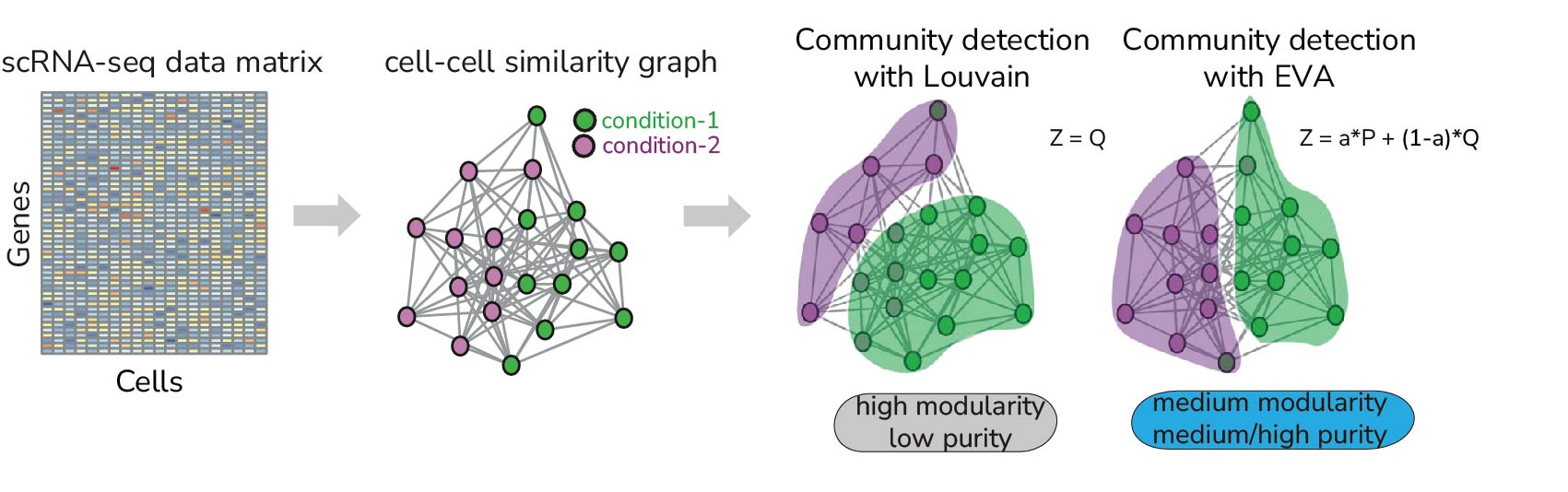
We have a long-term interest in using tools from network physics to address computational/statistical challenges in the omic data field. For instance, we recently explored using cell-attribute aware community detection algorithms to detect differential abundance of cell-types in scRNA-Seq data (ELVAR algorithm). We have also pioneered the application of spin-glass community detection algorithms to identify network module joint epigenetic-transcriptomic biomarkers (FEM algorithm) for early detection of esophageal adenocarcinoma. We continue to explore network theoretical concepts