Active Software
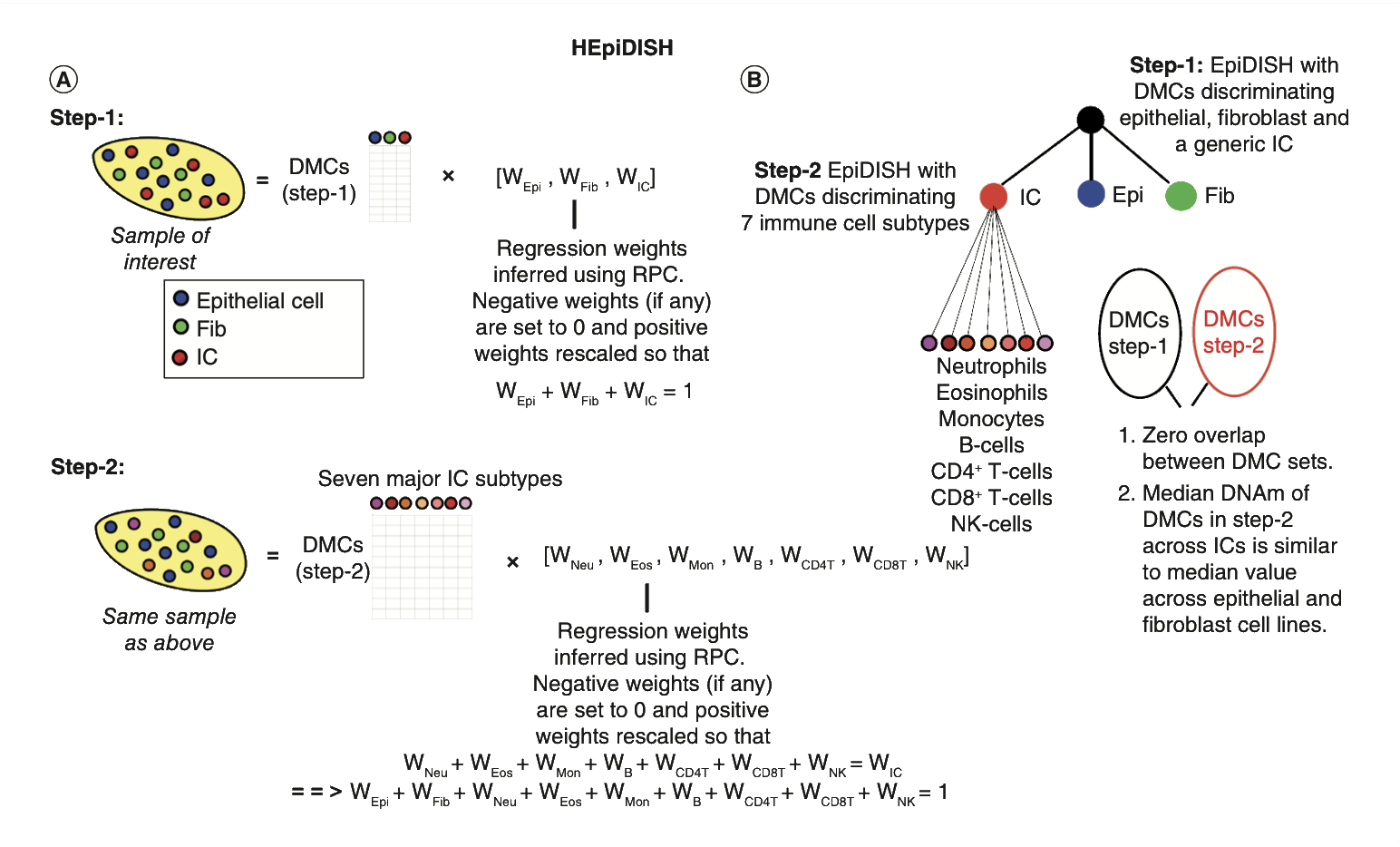
EpiDISH is a R-package to infer the proportions of a priori known cell-types present in a sample representing a mixture of such cell-types.
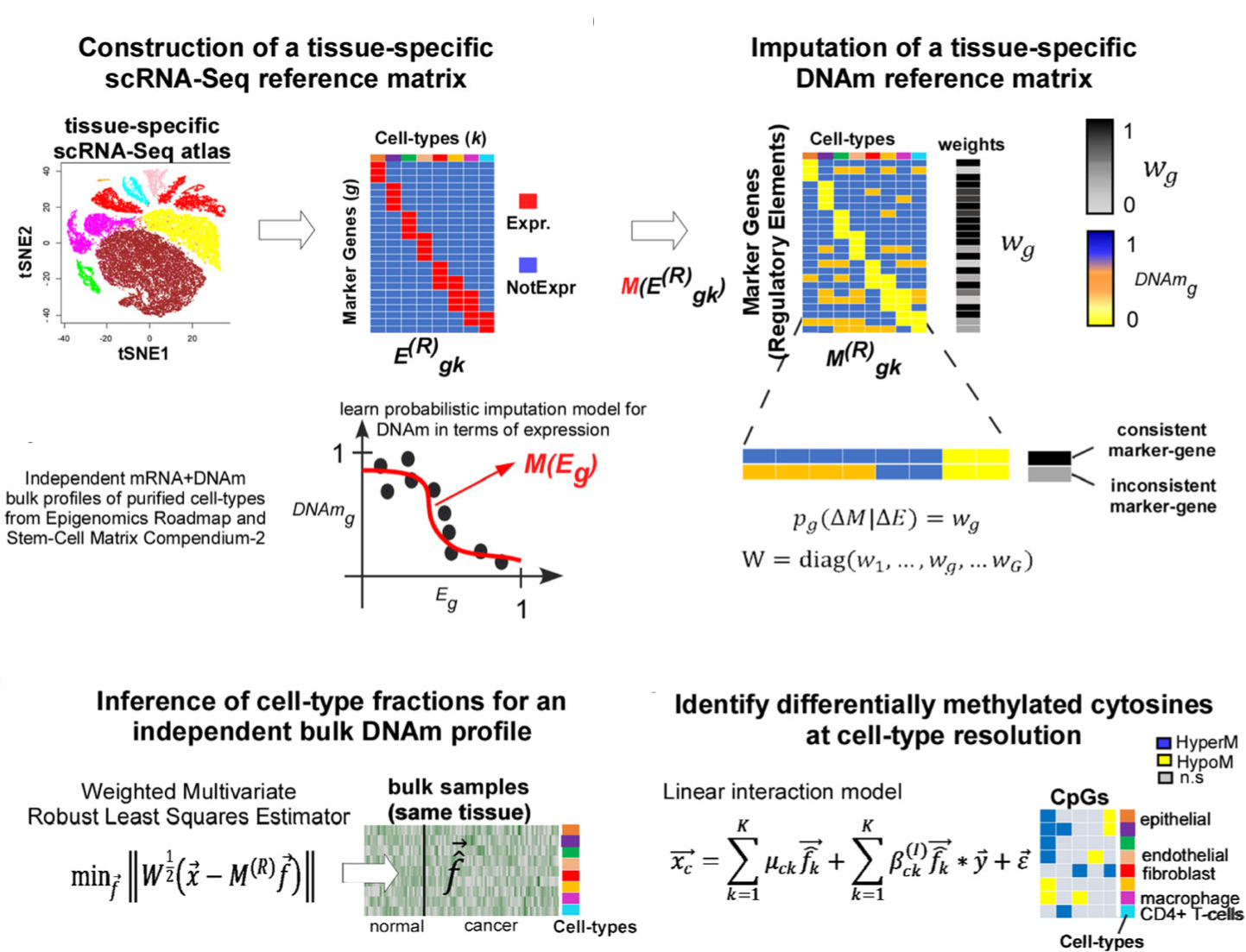
EpiSCORE is an R-package that leverages tissue-specific single-cell RNA-seq atlases to computationally impute DNA methylation reference matrices for complex solid tissues, enabling accurate reference-based cell-type deconvolution and cell-type–specific epigenetic analysis from bulk DNAm data.
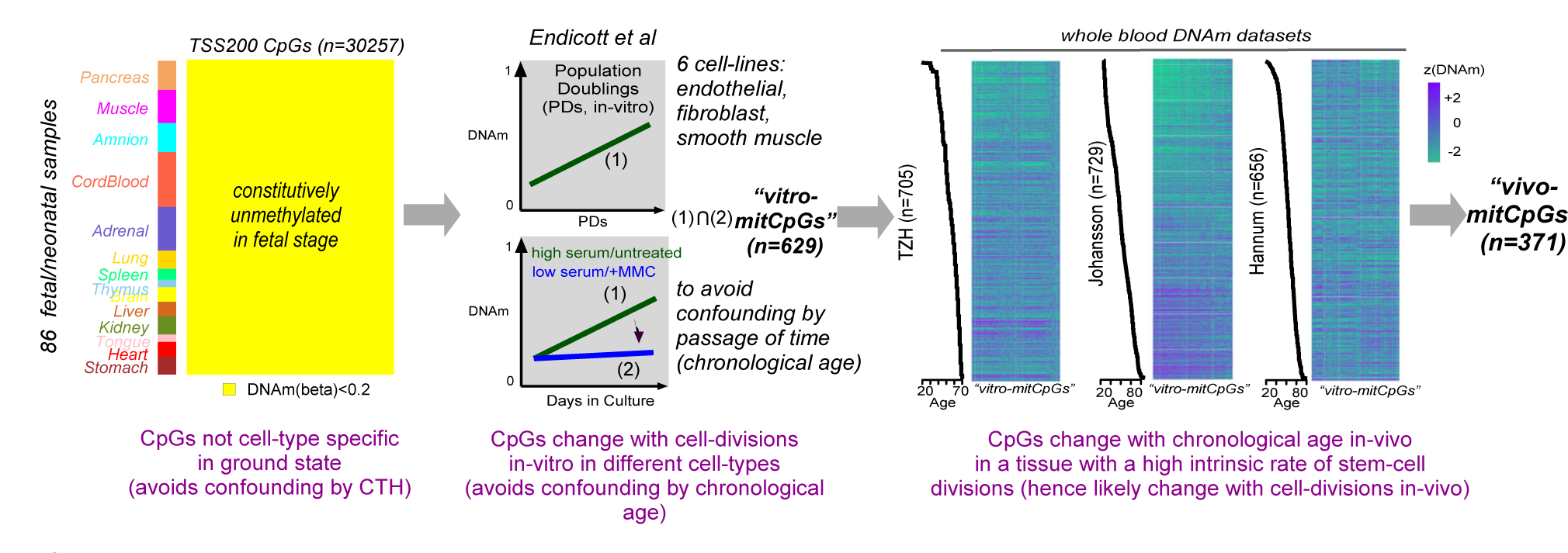
EpiMitClocks is an R-package to provide functions for estimating the mitotic age of tissues from a corresponding DNA methylation (DNAm) profile
SCENT is an R-package to provide a means of estimating the differentiation potency of single cells without the need to assume prior biological knowledge such as marker expression or timepoint.
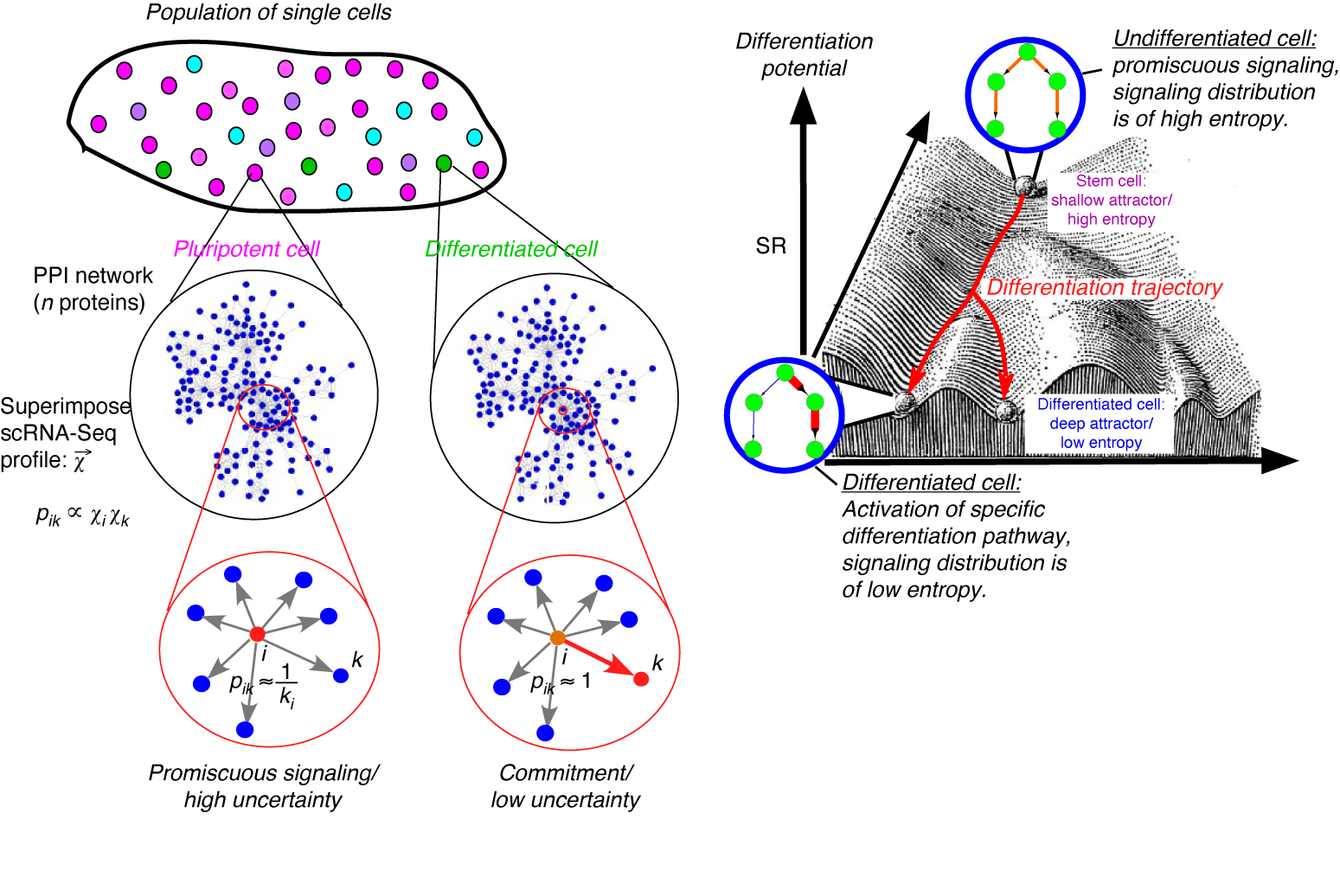
CancerStemID is an R package that uses single-cell RNA-seq data spanning normal to cancer stages to estimate measures of cancer risk and identify stem-like cancer cell populations.
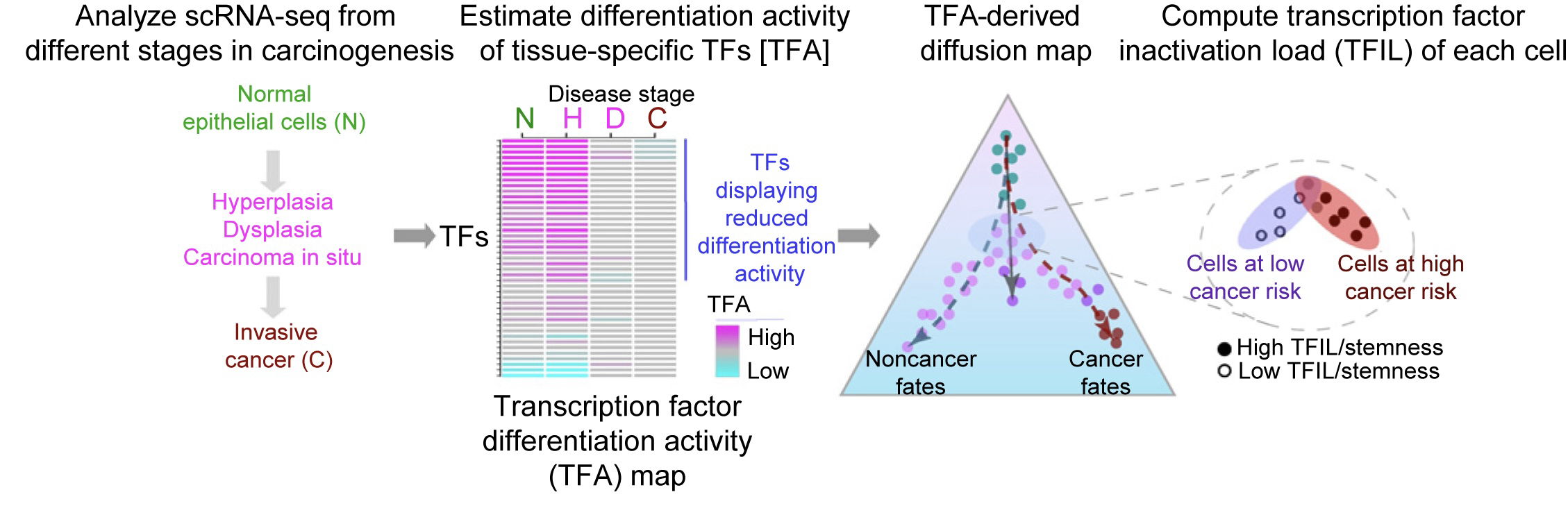
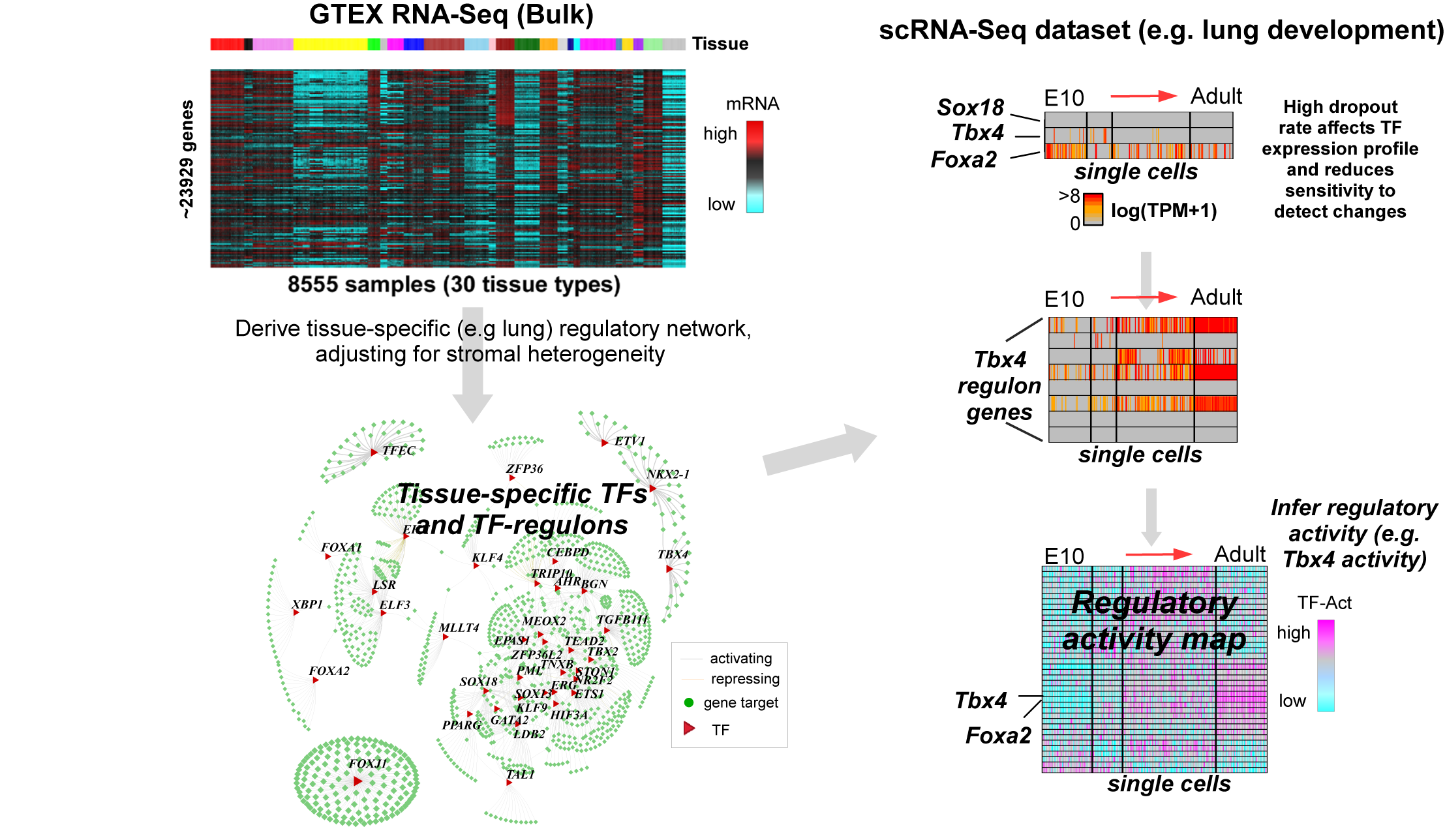
SCIRA is an R-package aimed at estimating regulatory differentiation activity of transcription factors in scRNA-Seq data.
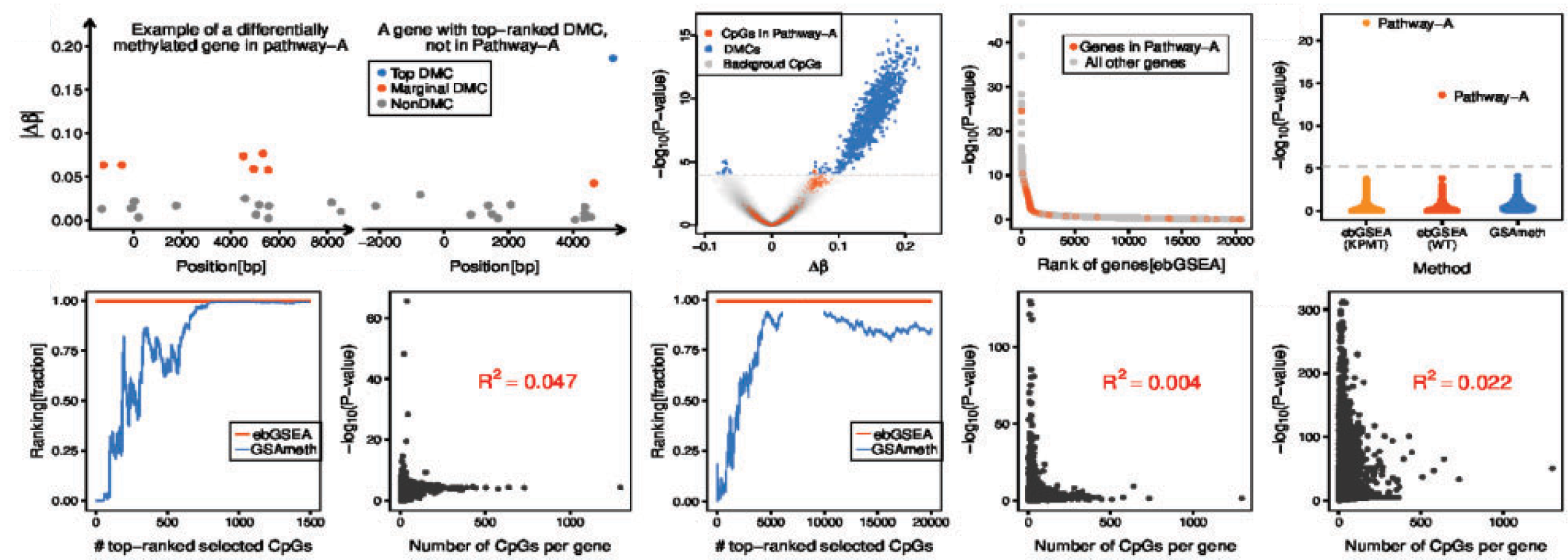
ebGSEA is an empirical Bayes gene-set enrichment method for EWAS that ranks genes—rather than individual CpGs—by their overall differential methylation, enabling unbiased and sensitive detection of enriched biological pathways.
ELVAR is an R-package for differential abundance (DA) testing of cell-types in single-cell RNA-Seq data.
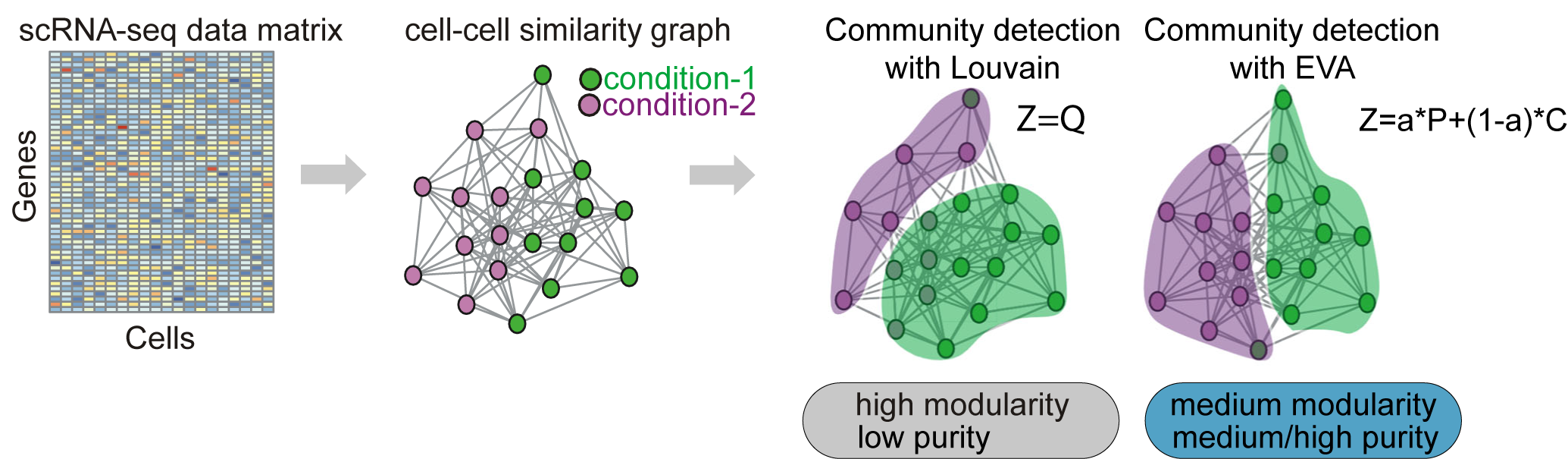
DICE is an R-package which uses the concept of Distance Covariance Entropy to help quantify bifurcation dynamics from scRNA-Seq data.
Legacy Software
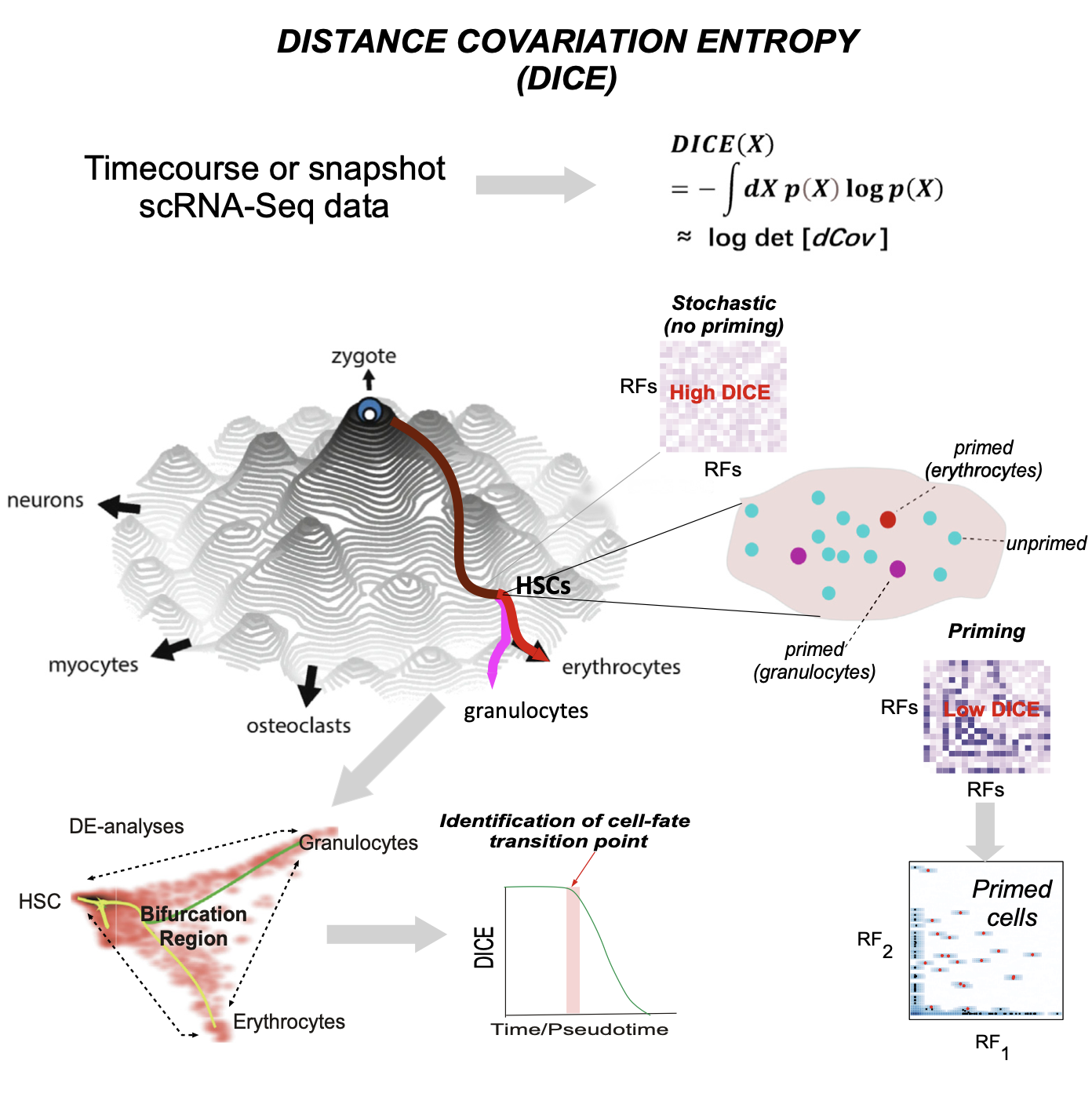
iEVORA is a DNA methylation–based algorithm that detects cancer risk markers by first identifying CpG sites with unusually high variability in at-risk tissues and then prioritizing those that also show changes in mean methylation.
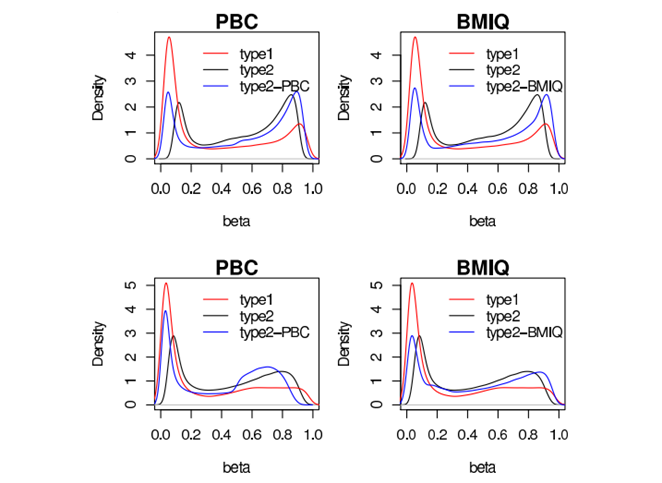
BMIQ is an R-package corrects Type-II probe bias in Illumina DNA methylation arrays by aligning their distributions to Type-I probes through a three-step beta-mixture–based adjustment.
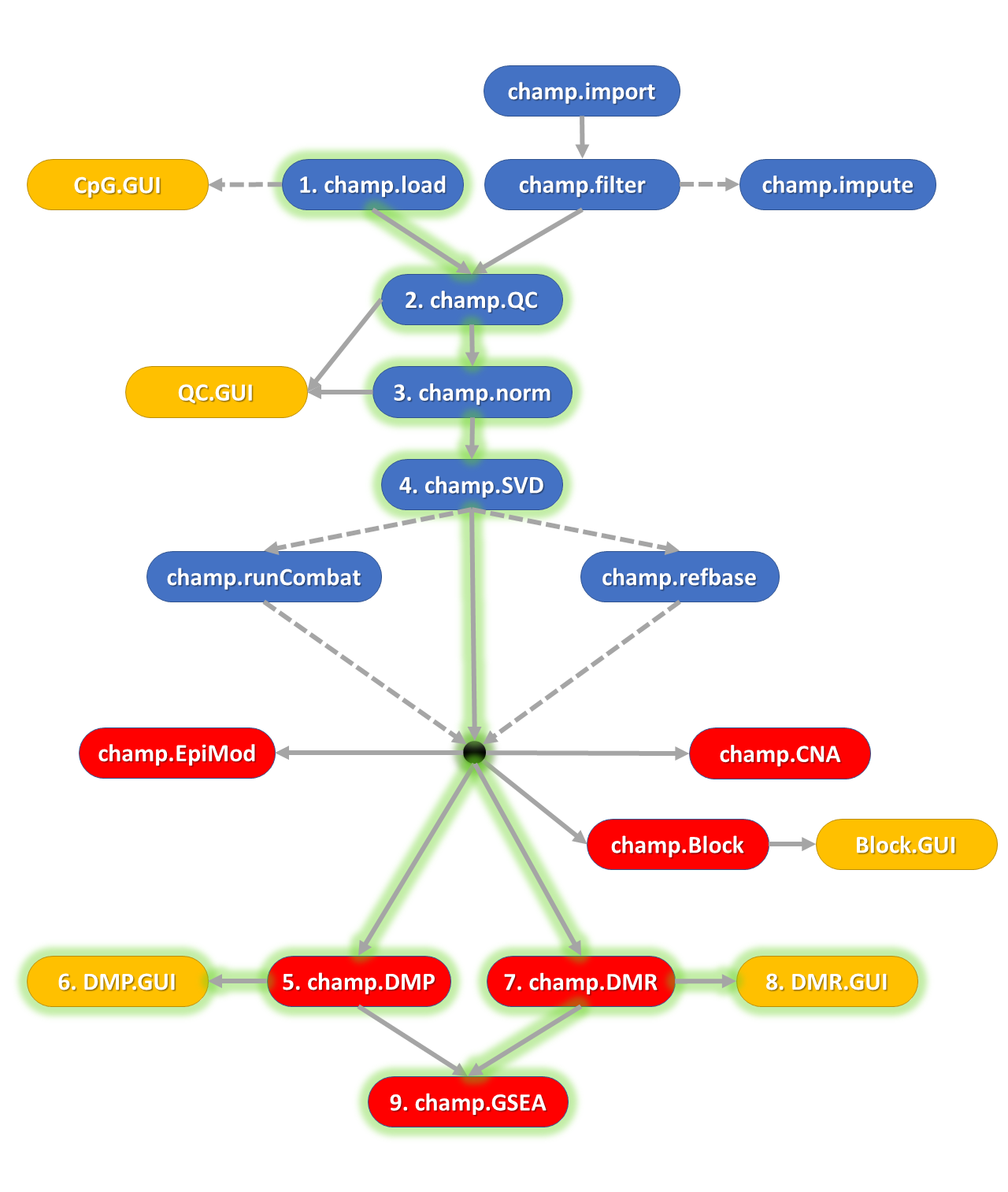
ChAMP is an R package that provides quality-control metrics, a range of normalization methods, and novel tools for identifying differentially methylated regions and detecting copy-number alterations.
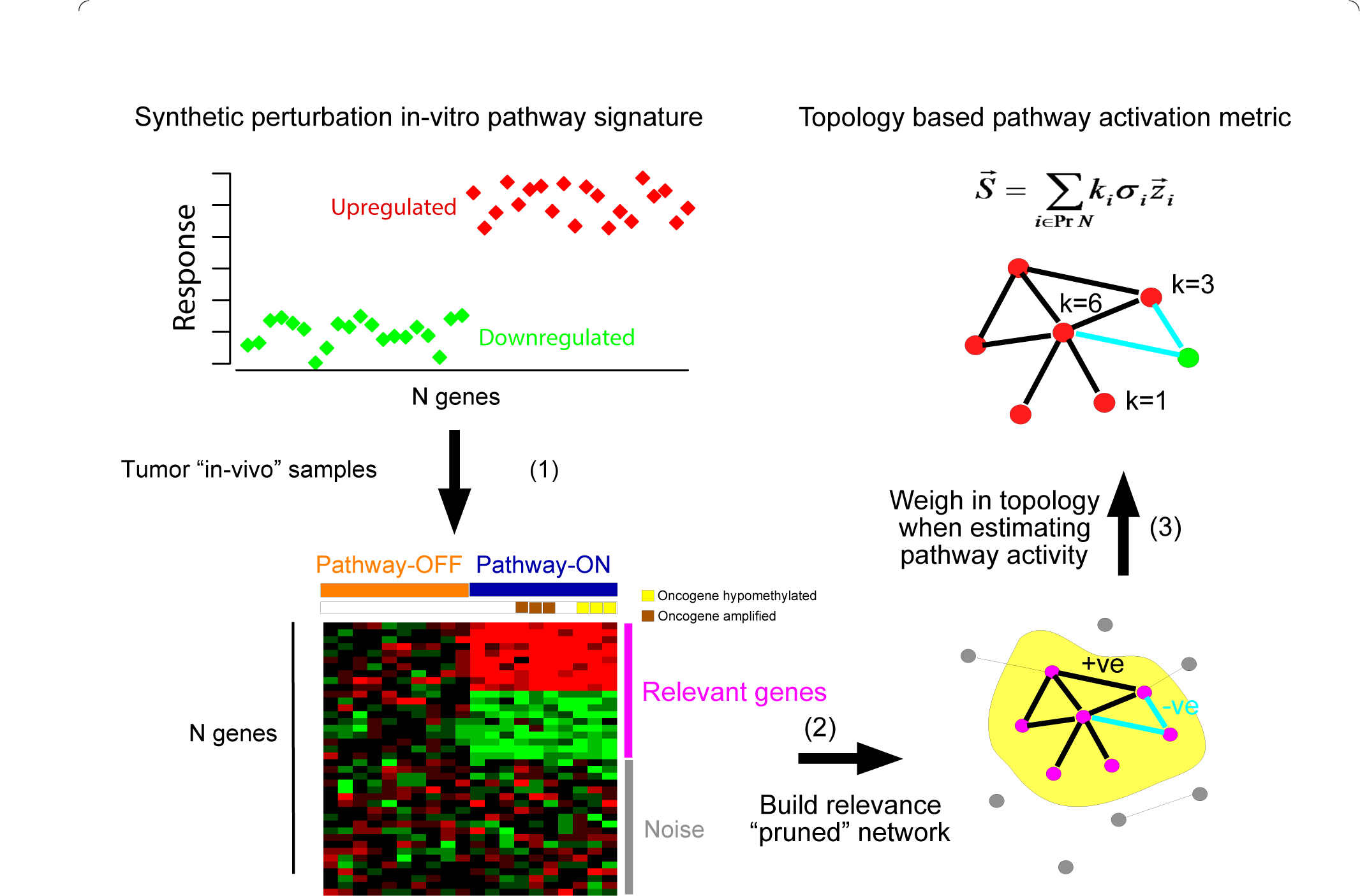
DART is an R package for evaluating the consistency of prior molecular signatures (e.g., in-vitro perturbation expression signatures) in independent molecular datasets (e.g., gene expression data).
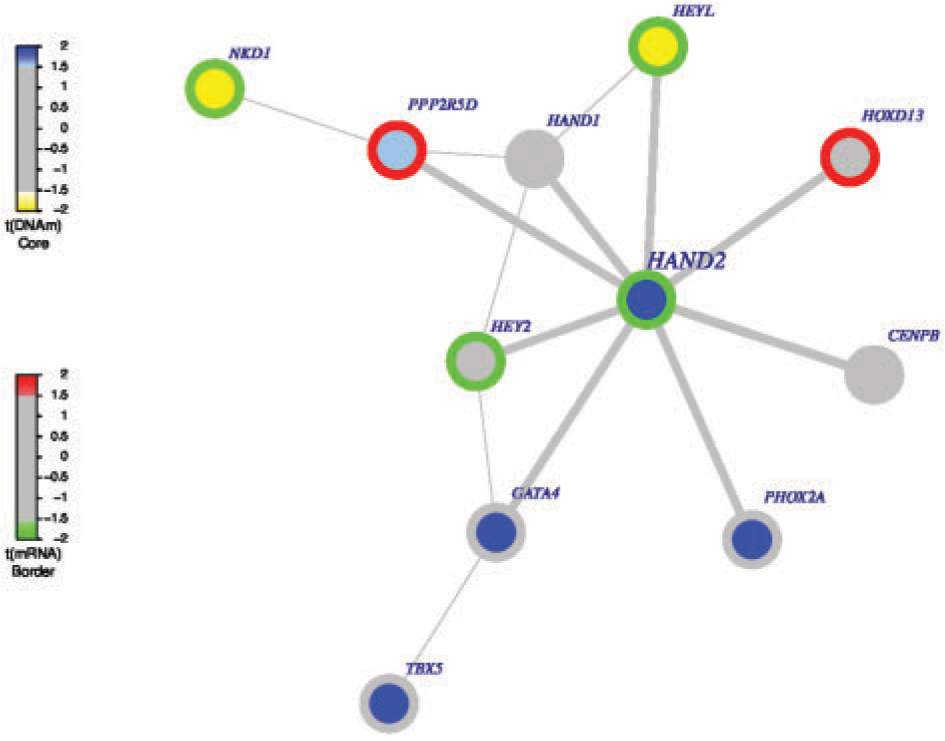
FEM is an R package for identifying gene modules showing coordinated differential expression and differential methylation associated with a phenotype of interest.
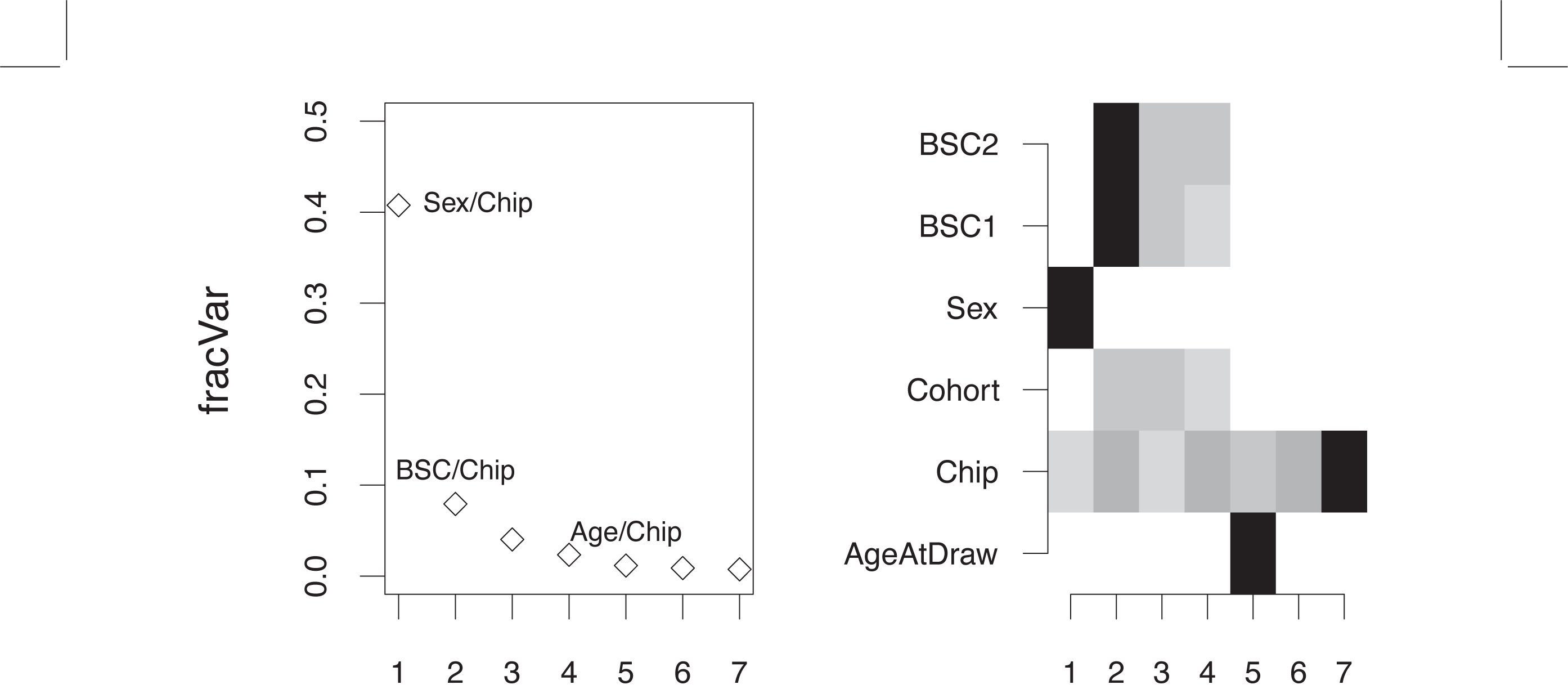
isva is an algorithm for feature selection in the presence of potential confounding factors
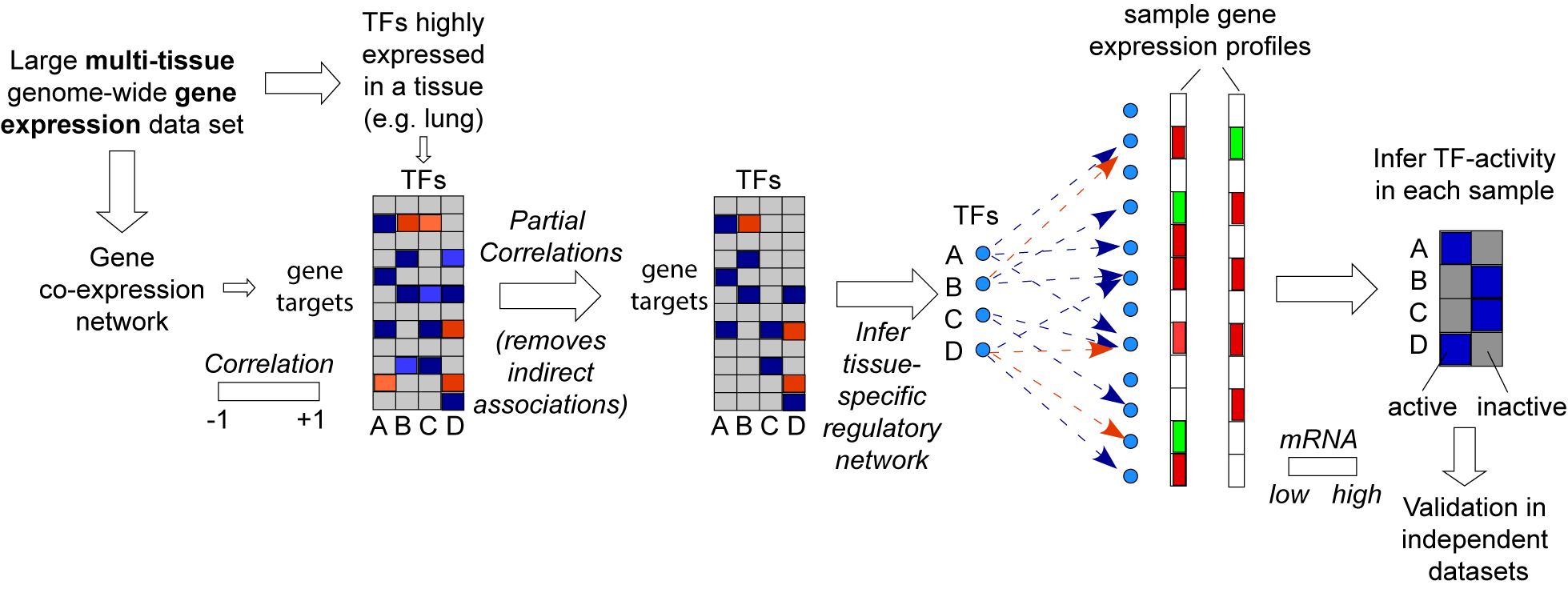
SEPIRA is a systems-epigenomics algorithm that builds tissue-specific regulatory networks from large gene-expression datasets to infer transcription factor activity in samples profiled by either RNA expression or DNA methylation.
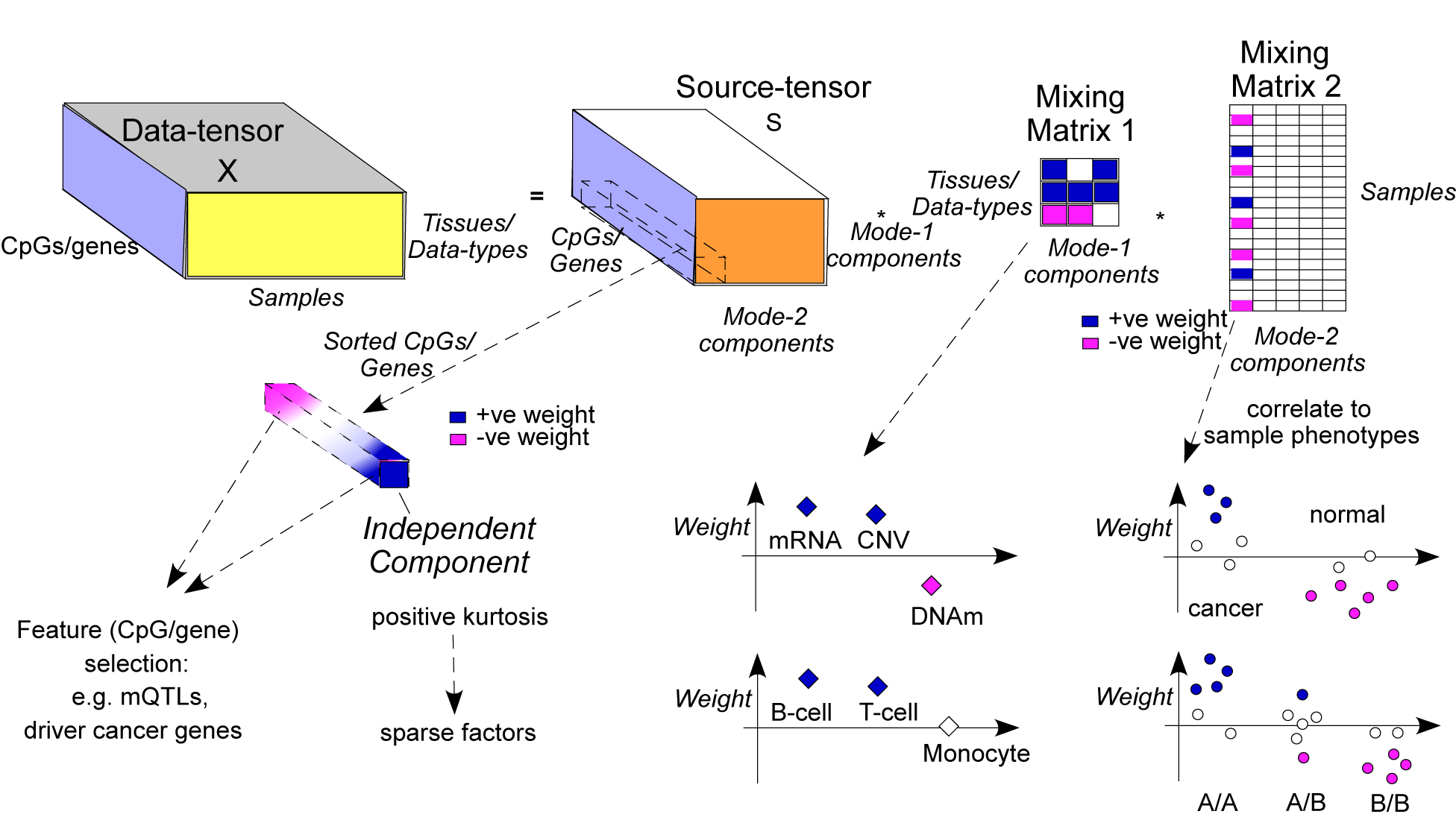
tensorICA is a tensor-based independent component analysis method that integrates multi-omic datasets to uncover shared biological variation, detect regulatory drivers, and identify disease-relevant molecular modules with high sensitivity and computational efficiency.