ChAMP (The Chip Analysis Methylation Pipeline)
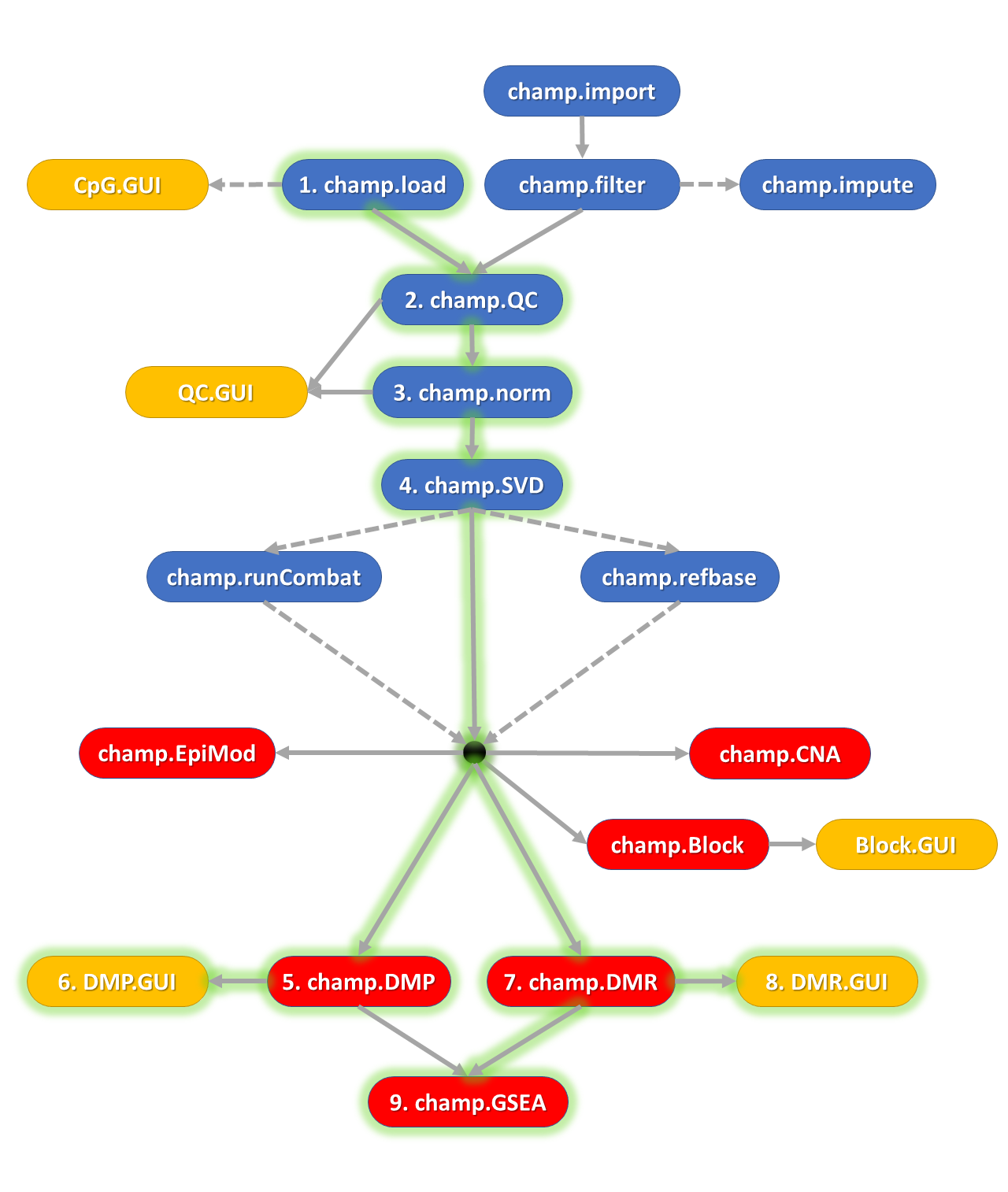
The ChAMP package is designed for the analysis of Illumina Methylation beadarray data (EPIC and 450k) and provides a pipeline that integrates currently available 450k and EPIC analysis methods. The new version of ChAMP extends and improves this analysis pipeline, adding novel and enhanced functionalities, including detection of differentially methylated genomic blocks (DMB), Gene Set Enrichment Analysis (GSEA), a variety of methods for correcting cell-type heterogeneity and detection of differentially methylated gene modules. Notably, the new package provides a series of web-based graphical user interfaces (GUIs), which facilitate analyses and enhance user-experience.
Download and Install
Citation (from within R, enter citation("ChAMP")):
- Tian Y, Morris TJ, Webster AP, Yang Z, Beck S, Feber A, Teschendorff AE. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 2017 Dec 15;33(24):3982-3984. https://doi.org/10.1093/bioinformatics/btx513
Maintainer: Yuan Tian <champ450k at gmail.com>
Installation
To install this package, start R and enter:
source("http://bioconductor.org/biocLite.R")
biocLite("ChAMP")Documentation
To view documentation for the version of this package installed in your system, start R and enter:
browseVignettes("ChAMP")HTML: ChAMP Tutorial